
3歲半的端端聽話地躺在對他而言較大的病床上,吊針扎進皮膚,身前貼著心電實時監測設備,儀器的線纜散落在床鋪一角。這一年多,他快速適應著每周打針的生活,已經不哭不鬧,爸爸說這是在輸入「能量」,他也選擇相信。
一年半前,來自浙江的端端確診罕見病黏多糖貯積症IVA型。由美國製藥公司Biomarin Pharmaceutical Inc.生產的「唯銘贊」,是目前唯一一種適用於該罕見病IVA型患者疾病緩解的藥物。
然而,明年5月,該藥品即將退出中國大陸市場。今年6月,藥企回應南都、N視頻記者稱,公司已決定不再為唯銘贊在中國的續期,目前的進口藥品註冊證將在2024年5月到期終止。
沒了藥,未來要怎麼辦?廣州患兒家長李曉麗最近總是在想這個問題。她迷茫地告訴南都記者,「如果停了藥,對我們來說,唯一可能的希望都沒有了。」
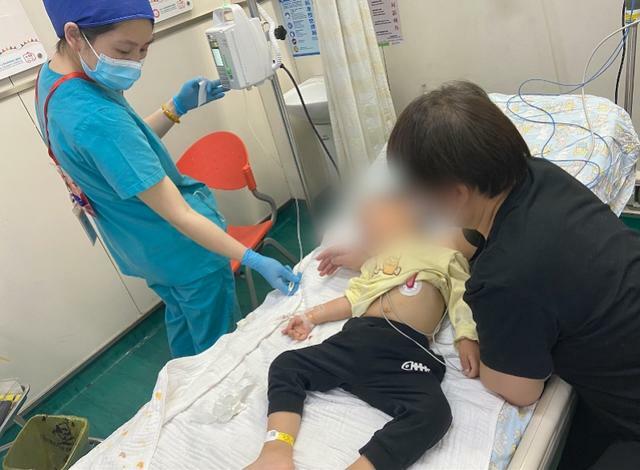
端端正在打針治療。
被困的成長
「我們很愛她,受到了很大的打擊。」李曉麗想不通,自己只是一個普通人,過著普普通通的生活,為何「這種像電視劇一樣的罕見劇情會發生在自己的孩子身上」。採訪時,她忍不住低泣著告訴南都記者,這幾乎是她人生中最黑暗的時刻,沒有比這更難接受的事情了。
大約一年前,李曉麗發現了女兒彤彤與同齡小朋友的區別,「她身高一直不是很高,運動能力也比較弱。」起初以為只是小問題,卻不料,在檢查後彤彤被確診了李曉麗一家此前從未聽聞的罕見病——黏多糖貯積症IVA型。
即使盡力治療,但這一年間,疾病依然在逐漸侵蝕著彤彤的幼小的身體:她的腿慢慢變形,走路依然不穩,脫掉衣服後能看到明顯的肋骨外翻……原本該上幼兒園的她,還沒有找到合適的學校。
患病後,彤彤的成長像是在「後退」。
「可能對於其他小朋友來說,在成長過程,他們的自理能力會慢慢增強,但她卻沒辦法做到。」面對孩子的困境,李曉麗時常也覺得痛苦又無力,「像她們這種孩子,骨骼比較脆弱,所以你雖然有意教她,她也有意學,但她確實力氣使不上來。」
李曉麗不希望孩子的人生被疾病困住、被退化的身體束縛,而是有質量地生活。
2019年10月,浙江人羅舟驚喜地迎來了孩子端端的出生。一歲半前,孩子都沒有異常,直到身高停止在83公分。與此同時,他發現孩子還有點脊柱後凸,總弓著腰,走路也一直晃晃悠悠。
檢查後,醫生告訴羅舟,端端不僅是脊柱後凸,還有髖關節發育不良。隨後的一長段時間裡,羅舟帶著兒子開始頻繁地前往醫院,但一直都只是做著骨科檢查和治療。他給孩子定製了兩個矯正支具,但孩子戴上後反而更容易重心不穩在走路時摔倒,其中一個治療脊柱的支具便閒置了下來。
半年後,端端的情況不見明顯好轉,羅舟又掛了浙大兒院的號,在醫生的建議下,帶著端端做了基因檢測,不久後便被確診黏多糖貯積症IVA型。「這個罕見病發展到最後很嚴重,終身的身高只有1米2左右,孩子上小學還能走路,初中可能就需要拄拐杖,往後可能要坐輪椅,甚至預期壽命也變短。」羅舟不敢想像,自進入患者群以來,群里已經有幾個大約九歲的患兒離開人世。
浙江大學醫學院附屬兒童醫院內分泌科主任醫師鄒朝春向南都記者介紹,黏多糖貯積症(MPS)是一組先天代謝異常的遺傳罕見病。患者體內缺乏溶酶體不能分解,令黏多糖在體內不斷沉積,會對患者全身骨骼、器官造成損害,甚至影響患者的壽命。
「最近發現了新的症狀,黏多糖貯積症目前應該要分8種類型,不同類型症狀會有差異。」鄒朝春向南都記者介紹道,患者剛出生時體內黏多糖堆積得還不多,因此看起來與常人無異,但隨著身體生長,走路慢、骨骼變形等症狀才顯現出來。
用患者家屬的話來說,黏多糖貯積症IVA型則是罕見病中的罕見型。酶替代治療成為大部分IVA型患者最合適的選擇,鄒朝春解釋,這種治療方法是通過藥物補充患者體內缺失的用於降解黏多糖的酶,起到緩解病症的作用。黏多糖貯積症IVA型目前的唯一適用藥物是唯銘贊(Vimizim, elosulfase alfa),但選擇用藥就是一輩子的事,患者終身都要靠藥物注射治療。
鄒朝春介紹,另一種療法是造血幹細胞移植,在有酶替代療法的前提下,大多數情況下不建議IVA型患者進行移植,可能有更大的風險。
唯一藥品即將退市
除了用藥,李曉麗不知道還能幫助孩子做些什麼,「很心痛,好像能為她做的不多。」
女兒彤彤確診後兩個月左右,用上了唯銘贊,藥物通過注射進入她幼小的身體。根據患者的體重,每五斤就需要一瓶5毫升的藥,每瓶藥7500元,每周注射一次。如今,彤彤一次需要用6瓶藥,每周打針就需要4.5萬元。
李曉麗估算,如果每周都用藥,一年下來,彤彤的藥費高達兩百多萬。她解釋,2022年剛開始用藥時,藥企有贈藥優惠,一年只要自費50萬,其他可以使用贈藥,但現在贈藥優惠也取消了。即使能通過「穗歲康」保險報銷一百萬,也還要自費一百餘萬。
彤彤斷斷續續地用藥,中途也曾因疫情停藥。每次去醫院用藥打點滴,都要花六七個小時。一整天,李曉麗都全神貫注在彤彤身上,「孩子打針其實不是很配合,用藥時又要看滴速。」
但藥物總算延緩了疾病的侵蝕,她感覺用藥後,孩子的體力會變好一些,雖說和同齡小孩依然有差距,但身高也不再停滯不前。
然而,不久後,李曉麗就聽到唯銘贊要退市的風聲。
許多IVA型患者家屬告訴南都記者,由美國製藥公司Biomarin Pharmaceutical Inc.生產的唯銘贊是目前唯一一種適用於MPS IVA患者疾病緩解的藥物。
「小朋友用藥本來就已經花了這麼多的精力和金錢,如果斷藥,就相當於前功盡棄,對他們會有很大的影響。」李曉麗每天都在想未來要怎麼辦,她迷茫地說,「如果停了藥,對我們來說,唯一可能的希望都沒有了。」
「讓孩子們用上藥」,也是正宇黏多糖罕見病關愛中心負責人鄭芋創立這一組織的初心。為讓唯銘贊進入中國大陸市場,她還曾追至美國。
鄭芋的女兒也是黏多糖貯積症IVA型患者,為給女兒求藥,她多年間四處奔波,得知美國有藥企研發了一款特效藥,便「飛」往美國。直到唯銘贊在美國獲批上市,她又努力接觸藥企商談,希望能讓國內的孩子也用上特效藥。
「一個人的力量太小了,沒有聲音。」鄭芋告訴南都記者,「我想,我們是不是也能團結到一起,共同面對這個疾病,向社會各界呼籲,引起重視,最後能有社會保障支持孩子們用上藥。」
南都記者查詢獲悉,2018年11月,國家藥品監督管理局藥品審評中心發布《關於發布第一批臨床急需境外新藥名單的通知》,唯銘贊正在名單中。據悉,該藥品於2014年首次在美國獲批上市。
2019年,鄭芋終於等來了唯銘贊,沒想到短短几年就要經歷藥品「退市」。「那時我以為已經熬過了我們最痛苦的那些年,但現在剛有一點希望,藥企又把我們拋棄了。」
今年6月,針對藥品退市問題,南都記者聯繫藥企進一步了解情況。BioMarin Pharmaceutical公司回應南都記者稱,公司已決定不再為唯銘贊在中國的進口藥品註冊證續期,目前的進口藥品註冊證將在2024年5月到期終止。
該公司稱,「複雜的市場准入架構令某些藥物供應難以為繼,特別是罕見病治療用藥。在過去幾年中我們盡了最大努力,對唯銘贊可行的報銷機會進行商討,但收效甚微,因此我們決定不再繼續註冊該產品的上市許可證。」此外,公司正探索各種方法,讓目前正在接受治療的患者能夠持續獲得藥物供應。
第二根「救命稻草」
「可行的報銷機會」也是家長正迫切尋求的。除了用藥,醫保是罕見病家庭的第二根「救命稻草」。
家在浙江的張鈺平是國內較早開始為孩子用藥的家長,為此不惜花費高昂的藥費,而讓唯銘贊進醫保,曾是她堅持用藥的重要理由之一。
2019年7月,張鈺平的孩子確診黏多糖貯積症IVA型,聽聞治療藥物唯銘贊同年已經在國內上市,價格雖高但很有可能進入浙江醫保,反覆權衡,她還是決定邊等待醫保傳來好消息,邊為孩子尋找其他治療途徑。
「那段時間,我的情緒很低迷,只要有時間就去醫院,北京、上海,中醫、西醫,我都帶孩子看過。不停在努力,不停在權衡,想過幹細胞移植、想過跳大神賭一下,最後都放棄了。」
張鈺平向南都記者回憶,直到2020年5月,她等來了唯銘贊未進入浙江醫保的消息,「我更絕望了,對孩子的自責愧疚就更多,因為覺得是父母沒有能力,沒有那麼多錢,才不能給她用上藥。」她想,「如果我有很多錢,不需要進醫保,一年200多萬對有些人來講也不算多。我很懊惱。」
目前,唯銘贊仍未納入國家醫保目錄。但各地已對罕見病用藥保障展開探索。
「有部分地區曾將黏多糖貯積症IVA型納入地方醫保,例如成都等。」北京病痛挑戰公益基金會信息研究總監郭晉川向南都記者介紹,例如,2021年,成都市醫療保障局發布《成都市罕見病用藥保障藥品範圍及認定標準》,唯銘贊被納入其中,享受罕見病用藥保障資金,在一個治療年度內累加計算、分段報銷。
郭晉川表示,但目前,各地的普惠型商業補充醫療保險才是這類罕見病的主要保障方式。他解釋,普惠保險各地報銷細則差異大,主要作用在於對醫保報銷的有效補充。
在廣東,一些城市的普惠型商業補充醫療保險已經為患者提供支持。廣州患者家長李曉麗告訴南都記者,通過「穗歲康」每年最高可以報銷100萬。
2023年,「深圳惠民保」將包括唯銘贊在內的7個罕見病自費藥納入保障,規定個人負擔費用年度累計1.6萬元以上部分,按照參保人連續參加深圳市重特大疾病補充醫療保險或「深圳惠民保」時間,分別支付50%、60%、70%,年支付限額50萬元。
但對許多罕見病家庭而言,在高額藥費之下,普惠型保險的幫助很有限。郭晉川認為,僅靠普惠型保險難以保證患者能持續規範用藥。一些地方的普惠型保險保額並不高,30萬到50萬左右,還存在既往症限制降低報銷比例,對於罕見病高值藥而言,保障水平有限,杯水車薪。
家長的自救
張鈺平想不能再等下去了,沒有醫保,她決定自己聯繫藥企降價。
2020年,她曾嘗試聯絡藥企的中國區工作人員,一談就是幾小時,給出一條一條的理由,力爭用藥的優惠。
「我說,中國這麼大的市場,企業通過快速審批通道就進來了,可見對罕見病病人的最大關懷。」她向南都記者複述著數年前的談判場景,「如果藥效特別好的話,我願意配合企業去真實地宣傳用藥效果。」她還向對方坦誠了自己和丈夫的工作,希望證明自己能給孩子好的照顧和引導,讓孩子成人成才,能對宣傳藥效起到好影響。
「那次,我應該還說了很多很多,談六七個小時。」
大約一周後,她爭取到了買一贈一的優惠,但仍要自費100多萬。張鈺平很掙扎,不知道這是不是一條能看到未來的出路,但那時咬咬牙還是選擇了用藥,「關鍵是我想我的孩子用上藥,如果有效果,爭取能為下次進醫保加籌碼。」經過幾年治療,目前張鈺平爭取到了一年自費50萬的價格。
張鈺平並非孤軍奮戰。
許多患者家長得知確診後都主動聯繫藥企,為孩子爭取藥品優惠。他們還嘗試聯繫基因治療公司等各方資源,諮詢出國治療等一切方法,希望發現一條新的出路。
2022年3月,來自浙江的端端開始打唯銘贊,每周需要5瓶,也就是37500元。「我算了算家裡的積蓄,決定不管能用多久,我們先給他用上。」權衡過後,羅舟很快決定要給孩子用藥,即使要終身使用,即使可能是個無底洞。
在高昂的醫藥費下,端端父親羅舟也嘗試聯繫藥企溝通,「藥方跟我說一年50萬自費,剩下的藥是可以贈送。」治療的58周,端端的藥費已經花了50多萬。
醫院裡,羅舟時刻盯著扎進端端手上的吊針,每隔15分鐘調一次滴速,每小時3毫升、6毫升……36毫升,由慢到快。端端也不再常因打針哭鬧,他的快速適應讓羅舟只覺得心酸,「以前他很抗拒的,每次我都說這是去輸能量,每輸一次就能量滿滿,可以隨便走,隨便跑了。」
正宇黏多糖罕見病關愛中心將這些同病相憐的家庭組織起來,同舟共濟,熬過今後的艱難時刻。「罕見病患者很少,很多家庭很迷茫,看不清未來怎麼辦。但病友如果有一個交流平台,可能在心理上就互相有一些慰藉。」鄭芋介紹,目前,加入關愛中心的患者已有600餘人,其中IVA型患者不足100人。

罕見病家庭參加線下義診活動。
患者家屬們成為關愛中心的志願者,幫忙組織線下義診、線上論壇等活動,越來越多醫學專家來到義診,現場為患者診治、分享現階段最新的治療手段、各地醫保及商保政策等。
絕望與希望
藥品即將退市的同時,贈藥也面臨被取消。今年2月,羅舟為端端申請到最後一批贈藥,只夠堅持到7月底。
「藥用完之後,再買的話都是全價,一年大概一百九十多萬。如果明年端端體重上漲了,要用6瓶藥,一年就是兩百多萬。」羅舟一筆筆算著帳,只能得出難以承擔的數字。「藥很貴,但是對我們來說,能在國內買到藥,起碼我們還有選擇。如果真的不賣了,我們就真的一點念想也沒了。」
「我設想過很多次未來。」羅舟清醒又絕望,「對我來說,我希望他能走在我前面,起碼我可以看著他走,是不是?」「他是我唯一的小孩,如果以後我死了,你說他要怎麼辦?誰能照顧他?他只要生活得開開心心就行,有時候,當父母的也只能這樣了。」
「退市」風聲放出時,剛確診的欣欣甚至還從未來得及用上藥。
3個月前,3歲半的欣欣確診黏多糖貯積症。父親徐樂陽給記者發來許多欣欣的照片:她穿著帶小亮片的紅色公主裙手拿玩具笑容天真,趴在桌前認真回答兒童繪本的問題,隔著玻璃觀看小朋友們學舞。

欣欣穿著帶小亮片的紅色公主裙。
照片中的她,看起來活潑快樂,好像不曾經歷病痛。但徐樂陽知道,孩子越長越大,病症也會越來越明顯。
「從小走路就會摔跟頭,後來也發現她的脖子有點短,手腕很鬆弛沒勁兒,還有一點雞胸。」徐樂陽告訴南都記者,受罕見病影響,孩子年初進行了扁桃體腺樣體肥大切除手術,甚至被檢查出聽力受損。
在短短數月內,徐樂陽跑遍了各地醫院,從對黏多糖貯積症一無所知,到半個專家。孩子的治療還需大筆費用,他與妻子也無法拋下工作,只能邊尋訪名醫,邊加班賺錢。他沒工夫想以後,只希望眼下能有辦法讓孩子治好病。
面對藥品退市,張鈺平則十分氣憤,「我不會放棄法律武器去維護我們孩子的利益。」她感受到一種「拋棄」,這是唯一一款藥,「我們為此支付了金錢、精力,企業不能說退就退了,這對病人們是不負責任的。」
氣憤、絕望後,張鈺平還是希望藥能留下,但並不是為了治癒病情,而是想為孩子爭取一個等待的時間。
「醫學發展也有探索過程,也許突破瓶頸就能有機會。」她抱著這樣的期望,「如果我這10年都給孩子用上目前最有效的治療方式,哪怕是代價再大一點,那麼10年之後,她可能就等來一個機會,讓她可以生活自理,可以發光發熱。」
電話掛斷前,張鈺平向南都記者坦言,「我查了很多資料,對這個病有一個客觀的認識,就是無法治癒。」
接著她又說,「但作為一個母親,我好像也沒什麼路可選了。」
(應受訪者要求,李曉麗、彤彤、羅舟、張鈺平、欣欣、徐樂陽均為化名)
出品:南都即時
采寫:南都記者 敖銀雪

























