前言
吲哚-3-甲醛類化合物是吲哚甲醯化的產物,是重要的精細化工原料,也是很多雜環化合物的支撐骨架,3-吲哚甲醛類化合物呈現出了廣泛的藥理活性,被應用於治療止吐、降壓、抗腫瘤、抗炎、抗抑鬱等諸多病症中。
在高壓條件下最高產率以達到90%,該反應設備要求高,環境污染大,對於雜環和芳胺等底物該反應產率較低。

實驗部分
主要儀器與試劑AvanceIII型400MHz核磁共振儀;CO-11-6-6型多模塊式碳化矽微通道反應器;HG-G1-A型製冷加熱控溫系統;AgilentTechnologies5973N型質譜儀。
吲哚、2-甲基吲哚、4-溴吲哚、4-甲基吲哚、5-溴吲哚、5-氯吲哚、5-碘吲哚、5-甲基吲哚、5-氨基吲哚、5-甲氧基吲哚、5-叔丁基吲哚、5-吲哚甲醛、5-乙醯基吲哚、5-硝基吲哚、7-氟吲哚、7-甲氧基吲哚。
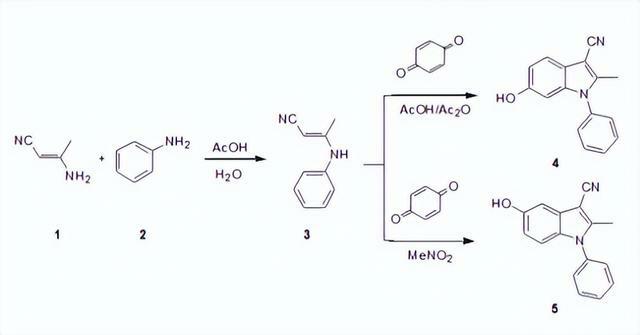
實驗室小試操作:50mL和10mL的Schlenk瓶放入磁子和橡皮塞,放入冰水浴中備用,向50mL的Schlenk瓶中加入(117.1mg)1mmol吲哚(1a),以及5.0mLDMF。
向10mL的Schlenk瓶中加入5.0mLDMF和(214.6mg)1.4mmolPOCl3,攪拌5min後用注射器將混合液體緩慢滴加到常溫下的50mL的Schlenk瓶中,然後反應溫度升至45℃攪拌反應1.5h,結束後把混合物倒入冰水中,接著加入0.4mol/L的氫氧化鈉溶液25mL並加熱到100℃,自然冷卻至室溫或放入冰箱中有白色結晶析出,抽濾,洗滌,乾燥,得到123.3mg淡黃色晶體,核磁表徵確認結構,產率為85%。

微通道反應器合成操作:首先向微通道中通入氮氣,設置溫度為25℃,接著向充入氮氣的500mL的三口燒瓶中加入0.3mol吲哚和300mLDMF,充分溶解後,依次接入流速為20mL/min的泵;向另外充滿氮氣的500mL三口燒瓶中先加入300mLDMF,在攪拌情況下加入0.48molPOCl3,然後接入20mL/min的泵。
準備工作完成後,開啟兩個泵,使物料流入微通道反應器內混合併反應,先通入2min使物料充滿微通道,然後計時開始,TLC檢測反應進度,物料在微通道反應器內停留時間54s,反應後的物料加入到500mL冰水中攪拌15分鐘淬滅,然後加入0.4mol/L的氫氧化鈉溶液煮沸後冷卻,結晶析出後進行抽濾,洗滌,烘乾稱重,得到39.2g淡黃色針狀結晶固體,產率90%。
實驗室小試操作同化合物2a,194.9mg淺紅色晶體,產率為87%,微通道反應器合成操作同化合物2a,28.2g淺紅色晶體,產率為84%,實驗室小試操作同化合物2a,145.5mg白色晶體,產率為81%,微通道反應器合成操作同化合物2a,20.4g白色晶體,產率76%。
實驗室小試操作同化合物2a,243.9mg淺黃色晶體,產率為90%,微通道反應器合成操作同化合物2a,34.5g淺黃色晶體,產率85%,實驗室小試操作同化合物2a,183.7mg白色晶體,產率為82%,微通道反應器合成操作同化合物2a,27.2g白色晶體,產率81%。
實驗室小試操作:50mL的Schlenk瓶中加入131.2mg(1mmol)2-甲基吲哚和5.0mLDMF,向10mL的Schlenk瓶中加入DMF5.0mL和214.6mg(1.4mmol)POCl3,攪拌5min後用注射器將混合液體緩慢滴加到常溫狀態下的50mL的Schlenk瓶中,加完後將反應溫度升至45℃,攪拌反應1.5h。
反應結束後混合物倒入冰水中,加入25mL(0.4mol/L)氫氧化鈉溶液,加熱到100℃,將反應瓶自然冷卻至室溫,濃縮旋蒸,用乙酸乙酯進行萃取,分液,乾燥,進行柱層析純化樣品,洗脫劑為V(石油醚):V(乙酸乙酯)=3:1,旋幹得到108.3mg白色固體,產率為68%。

微通道反應器合成操作:首先向微通道中通入氮氣,溫度設置為25℃,接著向充入氮氣的500mL的三口燒瓶中加入0.15mol2-甲基吲哚和300mLDMF,充分溶解後,依次接入流速為20mL/min的泵,向另外充滿氮氣的500mL三口燒瓶中先加入300mLDMF,在攪拌情況下加入0.24molPOCl3,然後接入20mL/min的泵。
準備工作完成後,開啟兩個泵,使物料流入微通道反應器內混合併反應,先通入2min使物料充滿微通道,然後計時開始,檢測反應程度,物料在微通道反應器內停留時間54s,反應後的物料攪拌5min後加入冰水進行淬滅反應,然後加入0.4mol/L氫氧化鈉溶液煮沸後冷卻至室溫。

濃縮旋蒸,用乙酸乙酯進行萃取,分液,乾燥,進行柱層析純化樣品,洗脫劑為V(石油醚):V(乙酸乙酯)=3:1,旋幹得到15.5g白色固體,產率65%。
對於吲哚-3-甲醛,嘗試了一次公斤級的生產,微通道儲料罐一個儲存4.0mol吲哚和2.0LDMF,另一個儲存罐放置6.4mol三氯氧磷和2.0LDMF,進料泵流速均為20mL/min,停留時間為54s,通完時間為100min,產量為0.5公斤,按照一個工作日8h來計算,一天可以生產2.4公斤。

合成條件篩選
針對實驗室小試條件和微通道反應器合成工藝中的反應時間、反應溫度、POCl3用量、溶劑等影響因素進行了優化。
實驗室小試條件:1.0mmol吲哚、1.4mmolPOCl3和8mLDMF,考察反應溫度對反應產率的影響,實驗結果見圖1。
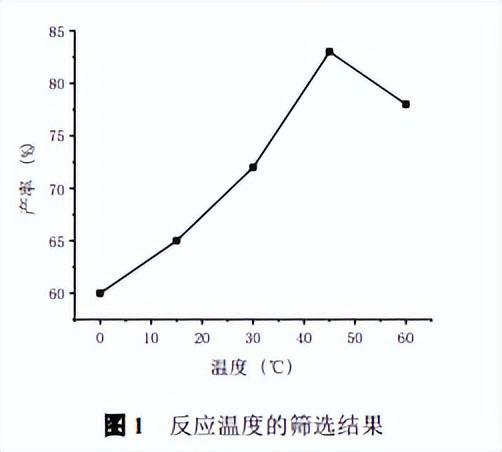
由圖可知,溫度對反應的影響比較大,0℃時由於反應溫度較低,反應物沒有完全反應,產率較低;隨著反應溫度的升高,在45℃時反應產率達到最高,繼續升溫副產物增加,產率反而下降,最終確定反應溫度為45℃。
實驗室小試條件:1.0mmol吲哚、1.4mmolPOCl3和8mLDMF,考察反應時間對反應產率的影響,實驗結果見圖2。

可以看出反應時間在1.5h的時候產率達到最大,繼續增加反應時間會有副產物產生,導致產率降低,確定反應時間為1.5h。
在1mmol吲哚和8mLDMF,溫度45℃,反應時間1.5h的條件下,考察了POCl3用量對反應的影響,實驗結果見圖3。

可以看出POCl3用量在1.2eq以下時吲哚有剩餘,轉化率和產率均較低;當POCl3用量增加到1.6eq時,吲哚完全轉化,繼續增加POCl3會浪費試劑和產生副產物,因此選擇POCl3用量為1.4eq。
在1mmol吲哚和1.4mmolPOCl3,溫度45℃,反應時間1.5h的條件下,考察了不同溶劑對反應的影響,反應結果見圖4。

發現二氯甲烷、乙酸乙酯作為溶劑時後處理過程繁瑣;四氫呋喃與水互溶,故產率並不是很高;DMF和乙醇同樣與水互溶,有意思的是使用DMF時產品可以結晶析出,因此選擇DMF以達到好的結晶效果。
綜上所述,通過篩選,優化反應條件為:DMF既作為反應溶劑也作為反應物,POCl3用量為1.4eq,溫度45℃,反應時間1.5h,可以達到比較理想的產率。
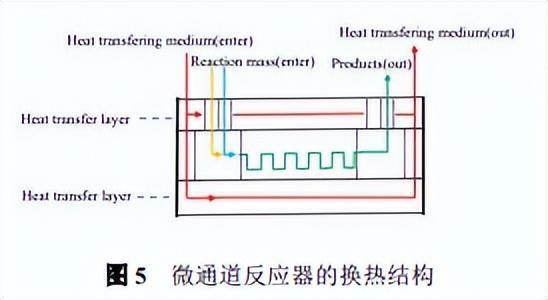
所使用的微通道反應器由進料泵、反應通道和製冷-加熱控溫系統組成,反應通道所用材料為碳化矽,反應器由微管路和外殼組成,製冷-加熱控溫系統對於反應通道的加熱和製冷通過導熱矽油完成,如圖5所示。
反應物料從進料泵進入反應通道混合反應,經過出料口從反應通道流出,所使用的微通道系統如下圖6所示。
①和②為反應原料儲存罐,①儲存DMF和吲哚,②儲存DMF和POCl3,原料準備好後由進料泵③和④(圖6)輸入到反應裝置⑤進行兩種物料混合併進行Vilsmeier-Hacck反應,反應完成後經導出管進入產品儲存器⑥,混合物再進行淬滅和結晶提純,產品經核磁表徵,微通道反應器的總容積為6×6=36mL。

從POCl3用量、微通道流速、反應物通過時間、反應溫度等因素來確定微通道反應器合成條件。
微通道反應器合成條件:0.15mol吲哚和400mLDMF,溫度25oC,進料泵的流速均為20mL/min,反應物在微通道內通過時間為54s,考察了POCl3用量對反應的影響,實驗結果見圖7。
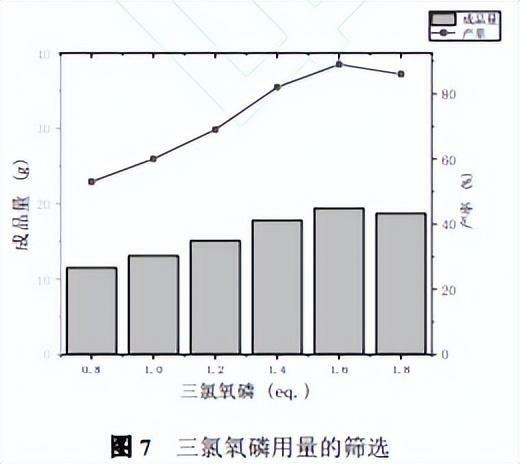
可以看出隨著POCl3用量的增加反應產率逐漸升高,達到1.6eq時吲哚轉化率為100%,同時產率達到最高,繼續增加到1.8eq時產率降低,造成POCl3浪費,同時配比太高會導致反應速率過快,造成微通道的堵塞以及生成其他的副產物,因此POCl3的用量為1.6eq。
通過調整進料泵的加料速度來控制停留時間,在0.15mol吲哚、400mLDMF和0.24molPOCl3,溫度25℃的條件下,考察了停留時間(加料速度)對反應的影響,實驗結果見表1。

隨著停留時間變長,產率從62%升高到91%,繼續降低加料速度停留時間過長反而產生副產物導致產率降低,因此加料速率確定為20mL/min,此時停留時間為54s,反應產率91%。
0.15mol吲哚,400mLDMF和0.24molPOCl3的條件下,反應物通過時間為54s,進料泵流速均為20mL/min,考察溫度對反應的影響,實驗結果見圖8。

發現從0℃升到25℃時產率逐漸升高,且均在80%以上,25℃時產率達到最高90%,繼續升溫反而導致副產物生成,說明溫度對反應的影響較小,這表明微通道反應器內反應物混合均勻,反應速率加快。
綜上所述,微通道反應器合成3-吲哚甲醛的條件為:POCl3用量1.6eq,溫度25℃,進料泵流速均為20mL/min,即通過時間為54s,DMF作為溶劑和反應物,通過對比實驗室小試和微通道合成,發現微通道反應器的生產效率大大提升。

達到同樣的產率小試要1.5h,微通道反應器則54s就能完成;在生產規模上,微通道反應器的36mL的容量反應1.5h可以得到百克產品,而50mL的Schlenk瓶反應1.5h只能完成毫克級別生產。
底物普適性研究
本文拓展了底物,考察不同取代基對該反應的影響。使用最小量0.15mol原料來進行反應,通過重結晶提純,其他產品採用柱層析進行純化處理,實驗結果如表2所示。

所有產物均進行了表徵,確認了化學組成,可以發現微通道反應器進行的克級規模生產,整體產率比實驗室小試條件下稍低,這是因為克級規模生產在進行後處理過程中產物結晶不完全導致的。
在14個衍生物中,不管苯環上取代基是吸電子的還是供電子的產率均在60%~90%之間,同時發現含有鹵素以及叔丁基的吲哚衍生物(化合物2a~2e、2l、2n)在後處理過程中都能正常結晶析出,而其他取代基衍生物需要柱層析純化。

可以發現吲哚甲醯化反應受底物取代基的電子效應影響比較明顯,當取代基為甲基(化合物2f~2h)、甲氧基(化合物2j~2k)、叔丁基(化合物2l)、氨基(化合物2i)等供電子取代基時,生成對應醛的產率較高,在69%~82%;當取代基為硝基(化合物2o),醛基(化合物2m)等吸電子取代基時,生成的醛的產率在68%左右;這是因為該反應屬於親電取代反應,供電子取代基增強了芳環上電子云密度導致產率增加。
由於鹵素在反應過程中的共軛供電子效應,穩定了反應中間體,因此含有鹵素(化合物2b~2e、2n)的衍生物產率都在77%~90%左右,對比化合物2f、2g、2h的產率,可以發現取代基的位置對反應也有影響,相同的基團在5位產率更高,這是由於取代基位阻小導致的。

結論
研究了Vilsmeier-Hacck反應合成吲哚3-甲醛類化合物,優化了微通道反應器合成的反應條件,與傳統間歇式反應釜式合成相比,微通道反應器合成時間縮短為54s,反應溫度從45℃降低到常溫,反應通量從毫克級到公斤級,該工藝適應性好,可以用於合成各類3-吲哚甲醛衍生物。