

克-雅病是一種罕見的致命性中樞神經系統退行性疾病。克-雅病臨床早期症狀不典型,症狀多樣且異質性較大,給該病早期識別和診斷帶來了很大困難。近日,中華醫學會神經病學分會神經感染性疾病與腦脊液細胞學學組全面分析國人克-雅病的臨床特徵和輔助檢查結果,同時結合國內外現有證據,編制了適合國人的克-雅病診斷指南——《克-雅病中國診斷指南2021》(以下簡稱指南)。
克-雅病既往又稱為亞急性海綿狀腦病或皮質-紋狀體-脊髓變性,按病因可分為散髮型克-雅病(sCJD)、遺傳型克-雅病(gCJD)、獲得型克-雅病(包括醫源型克-雅病及變異型克-雅病)。其中sCJD最為常見,約占85%;gCJD在同系血緣親屬中具有聚集發病現象,其確診依賴朊蛋白基因(PRNP)檢測出特定致病位點突變,占5%~15%;其餘為獲得型克-雅病。
在報導的中國人群中,克-雅病可見於成人各年齡段(18~87歲),中位年齡60歲左右,好發於50~70歲,尚無兒童發病的報導;克-雅病無性別傾向,男女發病比例相近。克-雅病多呈亞急性起病,也可以急性起病,呈卒中樣發作,疾病快速進展。一般無明顯誘發因素,其典型臨床症狀為快速進展性痴呆,同時伴有共濟失調、錐體系及錐體外系受累症狀、肌陣攣、視覺障礙等一系列症狀群。該疾病不可治,患者常在數月內死亡。
臨床表現
克-雅病患者臨床表現具有顯著異質性,即使在同一家系相同致病位點所致的gCJD患者也存在個體間差異。指南基於所有類型克-雅病患者共性和差異性的臨床表現,將其分為典型臨床症狀和非典型臨床症狀。
一、典型臨床症狀
1. 皮質受累症狀
(1)認知障礙:是克-雅病患者最常見的臨床表現。患者早期常表現為記憶力減退,判斷力、注意力下降等,隨疾病進展多數患者在數月內進展為痴呆。快速進展性痴呆是克-雅病患者最常見的特徵性症狀。至疾病晚期,患者可表現為無動性緘默、去皮質強直等。
(2)肌陣攣:肌陣攣是克-雅病的特徵性表現之一,尤其是聲光或皮膚觸碰誘發的肌陣攣,但在疾病早期或晚期如痴呆症狀較為明顯時,可無肌陣攣。
(3)精神症狀:發病初期可有輕微的精神異常,如情感淡漠或興趣下降,但仍保持相對正常的社會功能。隨著疾病的進展,患者逐漸出現如抑鬱、焦慮、易激惹、人格改變、脫抑制、幻覺、妄想等精神症狀。
(4)視覺障礙:表現為視力下降或視物模糊、視野缺損、視物變形(如視物顯小/大症、色覺障礙等)、視物成雙、皮質盲、Anton綜合徵等。部分克-雅病患者在疾病早期僅表現為孤立性視覺症狀,在隨後的幾周至幾個月內出現其他典型症狀。
(5)癇性發作:常見的發作形式包括局灶性運動性發作和全面性發作,多於疾病晚期出現。也有極少部分患者以持續性部分性癲癇或非驚厥癲癇持續狀態為主要症狀。
2. 小腦受累症狀
患者常表現為行走不穩,體格檢查可見共濟失調和眼球震顫。少部分患者可表現為孤立性共濟失調,至疾病晚期才出現認知障礙及其他症狀。
3. 錐體外系症狀
患者可表現為動作遲緩、肢體震顫和肌強直。在國人克-雅病患者中,以肌強直最常見,其他依次為運動遲緩、肢體震顫。
4. 錐體系症狀
大多數患者會出現皮質脊髓束受累的徵象,包括反射亢進、病理征陽性和痙攣等表現。
二、非典型臨床症狀
克-雅病的非典型症狀包括言語障礙,頭暈,頭痛,睡眠障礙(如嗜睡、失眠),肢體麻木或無力,自主神經功能障礙,肌萎縮,假性延髓麻痹,腦神經病變(如動眼神經、三叉神經、前庭窩神經損害),周圍神經病變,肌張力障礙(如舞蹈症、眼瞼痙攣、手足徐動症)等。
約30%左右克-雅病患者以非典型臨床症狀作為首發症狀,其中以頭暈和睡眠障礙最為常見。部分克-雅病可模仿其他中樞神經系統退行性改變,症狀類似於阿爾茨海默病、亨廷頓病、額顳葉痴呆、皮質基底節變性和進行性核上性眼肌麻痹等。
國人克-雅病患者的典型和非典型症狀及發生率見表1。
表1 國人克-雅病患者的臨床症狀發生率
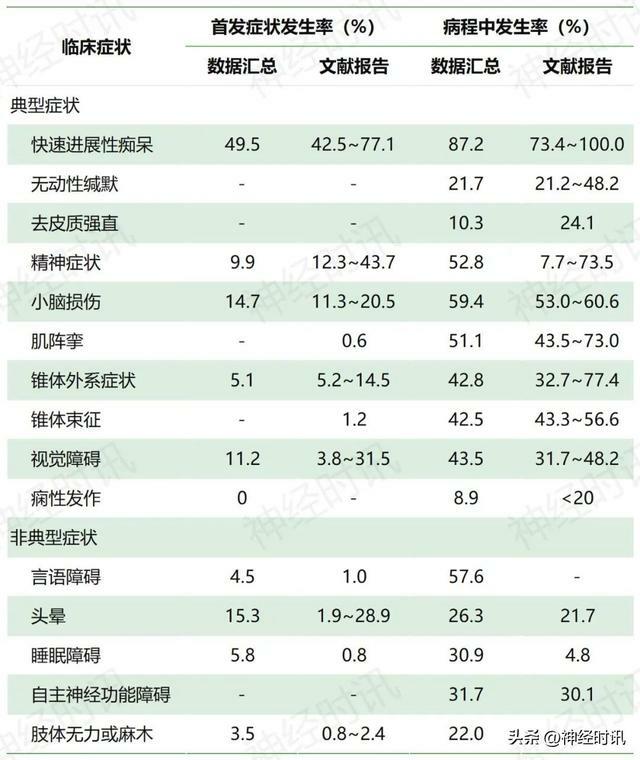
註:-:無或未見報導
推薦意見
(1)快速進展性痴呆是國人克-雅病最常見臨床症狀,當患者出現此症狀時,應考慮對該病進行排查;
(2)少部分克-雅病患者以非典型症狀為首發表現,此時輔助檢查可能有助於疾病早期識別;
(3)對懷疑克-雅病的患者應對上述皮質、小腦、錐體系和錐體外系及非特異性相關症狀和體徵行無遺漏詢問和體格檢查。
輔助檢查
一、實驗室檢查
現有證據表明,血液學檢查對於克-雅病的診斷價值有限,但腦脊液檢查對於克-雅病診斷和鑑別診斷具有重要意義。
推薦意見
(1)推薦所有懷疑克-雅病診斷的患者,常規行腦脊液14-3-3蛋白檢測;
(3)血和腦脊液總tau蛋白、磷酸化tau蛋白/總tau蛋白比值有助於克-雅病的診斷及鑑別診斷。
二、腦電圖
腦電圖可以為克-雅病的診斷提供較可靠的依據。在疾病初期,腦電圖常表現為基本節律的慢化;在疾病終末期,表現為低平腦電圖活動或α樣波。典型腦電圖表現為周期性尖慢複合波(PSWCs;圖1),多在疾病中晚期出現。這種波形具有以下特徵:每個綜合波持續100~600 ms,短周期(期間間隔0.5~2.0 s,約1 s 1次),且至少有5個重複的綜合波(每個綜合波時程差別需<500 ms)。
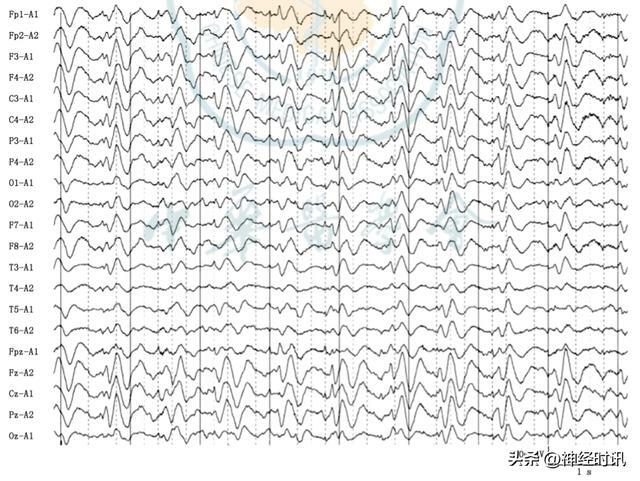
註:腦電圖可見雙側瀰漫性較多量低-中波幅三相波,雙側前頭部著
推薦意見
推薦所有懷疑克-雅病診斷的患者,常規行腦電圖檢查(不包括蝶骨電極和同步針極肌電圖),但PSWCs僅在病程中特定時期出現,在疾病早期及終末期少見。
三、影像學
頭顱MRI是診斷克-雅病的重要手段之一,絕大多數CJD患者可觀察到特徵性改變,即彌散加權成像(DWI)或液體衰減反轉恢復序列(FLAIR)上出現至少兩個皮質區域(額、顳、頂、枕;圖2A)和(或)基底節區[尾狀核和(或)殼核]高信號(圖2B)。
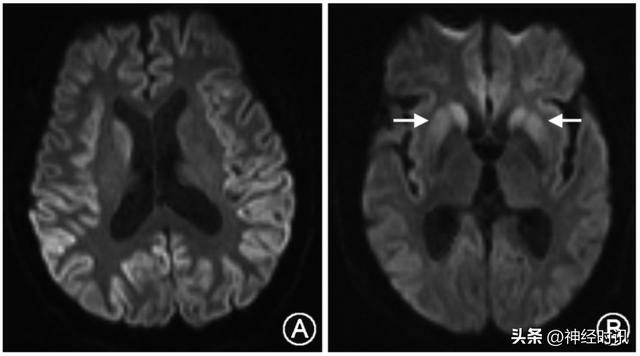
A:雙側額、顳、頂、枕葉皮 質及右側尾狀核頭彌散加權成像(DWI)高信號;B:雙側額、顳、 枕葉及尾狀核頭和殼核 DWI 高信號(箭頭)
推薦意見
(1)推薦對所有懷疑克-雅病診斷的患者,常規行頭顱MRI DWI/FLAIR檢查;
(2)建議有條件的醫療機構,行單光子發射計算機體層攝影(SPECT)或18F-氟代脫氧葡萄糖(FDG)正電子發射體層攝影(PET)檢查以協助診斷及鑑別診斷。
四、PRNP檢測
推薦意見
推薦對所有懷疑克-雅病診斷的患者,常規行PRNP檢測。
五、組織病理學
推薦意見
不推薦常規應用腦活檢用於CJD的診斷。
診斷
指南將克-雅病診斷分為3種級別:可能的CJD、很可能的CJD及確診的CJD(表2)。
表2 克-雅病的診斷標準

註:RT-QuIC:實時震動誘導蛋白擴增;DWI:彌散加權成像;FLAIR:液體衰減反轉恢復序列;PrPSc:致病型朊蛋白;PRNP:朊蛋白病基因;sCJD:散髮型克-雅病;gCJD:遺傳型克-雅病
鑑別診斷
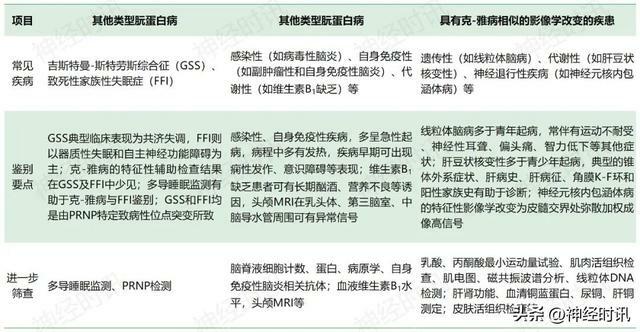
由於克-雅病臨床表現異質性較大,且諸多疾病可表現出與克-雅病相似的臨床特徵及影像學改變(表3)。因此,在診斷克-雅病之前,需進行詳細的排查,除外可治療性疾病。
表3 克-雅病的鑑別診斷
神經時訊整理自:中華醫學會神經病學分會神經感染性疾病與腦脊液細胞學學組. 克-雅病中國診斷指南2021 [J] . 中華神經科雜誌, 2022, 55(11) : 1215-1224. DOI: 10.3760/cma.j.cn113694-20220303-00151.
編輯 | 董曉慧 校對 | 譚靜妮

























